
Researchers Identify a New Therapeutic Target for Glioblastoma and Other Cancers

UCSF researchers have discovered that STAG2 – a gene commonly mutated in several human cancers – plays an essential role in DNA replication, revealing potential mechanisms for therapeutically targeting glioblastoma and other cancers.
As part of a multi-protein complex called cohesin, the STAG2 protein has been implicated in numerous cellular functions. The cohesin complex, which forms a ring-like structure that encircles DNA molecules, was first identified for its role in regulating the separation of DNA chromatids during mitosis (the process of cell division).
Published today in Nature Communications,1 UCSF neuropathologist and cancer biologist David Solomon, MD, PhD and colleagues identify a new role for STAG2. “We found that STAG2 is required for replication fork progression, which can be exploited as a therapeutic vulnerability in STAG2-mutant cancers,” said Solomon.
The replication fork is a structure that forms when DNA molecules are being replicated. Each strand of the DNA double helix is unwound and becomes a template for a new, matching partner strand, which is built and extended as the replication fork progresses.
The Solomon Lab reports that loss of STAG2, in normal cells, causes the replication fork to stall by disrupting interactions between the cohesin complex and DNA replication proteins. This leads to replication fork collapse and causes double-strand breaks in the DNA; consequent activation of DNA damage signaling pathways ultimately halts the cell cycle, thus preventing cell proliferation.
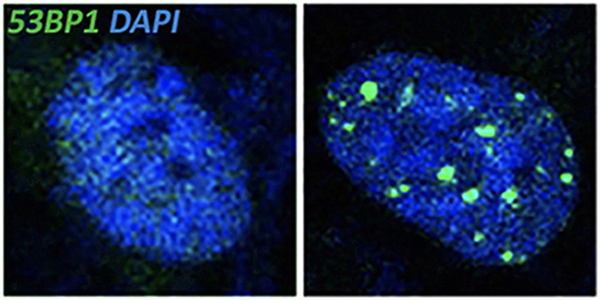
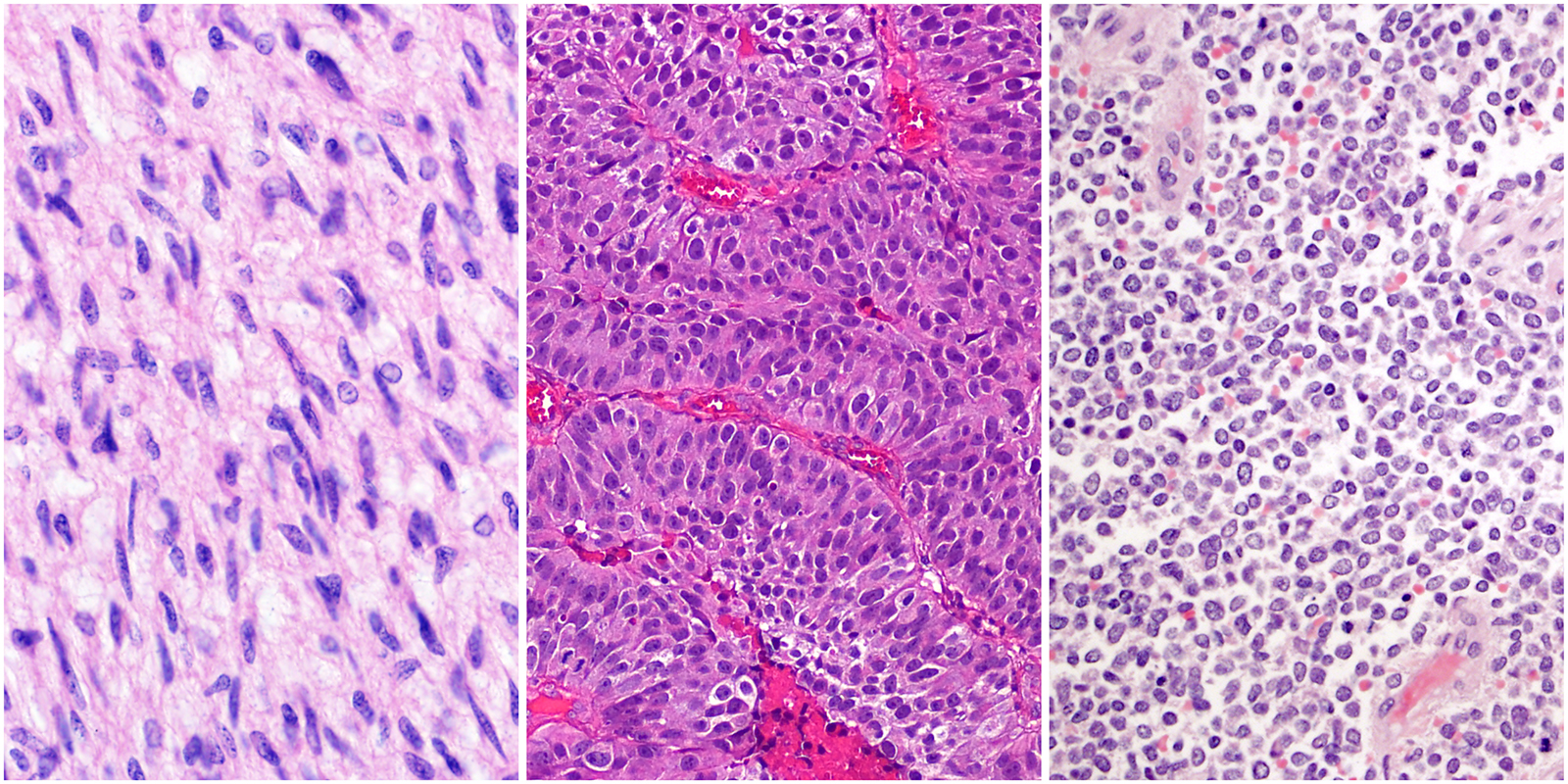
However, STAG2 inactivation behaves differently in tumor cells. “STAG2 inactivation is often associated with TP53 mutations, and in some cancers such as Ewing sarcoma, the combination of these mutations is associated with worse prognosis,” said Solomon. When they tested the combined loss of STAG2 and p53, those cells escape cell cycle arrest. Instead, the DNA double-strand breaks caused by STAG2 deficiency likely contributes to further gene mutations and rearrangements that promote tumorigenesis.
Given the accumulation of DNA double-strand breaks with STAG2 inactivation, Solomon and colleagues speculated that STAG2-deficient cancer cells might heavily rely on DNA damage repair proteins for survival. Indeed, the combined loss of STAG2 and certain DNA repair factors (e.g. ATR, PARP1, BRCA1) results in synthetic lethality, a condition in which the combination of two or more deficiencies leads to cell death.
When testing a panel of chemotherapy drugs, Solomon also found that STAG2-mutant cancer cells (including glioblastoma and Ewing sarcoma) are more sensitive to cytotoxic chemotherapeutic agents that induce double-strand breaks. They also have lower survival rates when exposed to gamma radiation. Presumably, loss of STAG2 sensitizes tumor cells such that an even greater accumulation of DNA damage is catastrophic.
“It’s very encouraging to find a targetable synthetic lethality for STAG2-mutant cancers,” says Solomon, “Increasingly, there’s a shift towards treating cancers using a precision medicine approach by matching specific targeted therapies based on their exact genetic mutations. This work lays the foundation for such targeted therapies for STAG2-mutant cancers.”
REFERENCES
1. Mondal G, Stevers M, Good, B, Ashworth A, and Solomon DA. (2019) A requirement for STAG2 in replication fork progression creates a targetable synthetic lethality in cohesin-mutant cancers. Nat Commun 10. doi: 10.1038/s41467-019-09659-z.
2. Solomon DA, Kim T, Diaz-Martinez LA, Fair J, Elkahloun AG, Harris BT, Toretsky JA, Rosenberg SA, Shukla N, Ladanyi M, Samuels Y, James CD, Yu H, Kim JS, Waldman T. (2011) Mutational inactivation of STAG2 causes aneuploidy in human cancer. Science 333(6045):1039-43.